Hello, this is the webSDA documentation.
Introduction
Simulation of Diffusional Assosiation (SDA) offers solutions to a number of simulation tasks for biological macromolecules and can reproduce experimental results. However, it cannot be used by a wide audience because of its multitude of different parameters and options that are often interdependent. Setting these parameters correctly and understanding their function requires expert knowledge in Brownian dynamics simulation. In addition, the program relies in part on shell scripts which limits it to Linux computers. webSDA is a web server which aims to eliminate these obstacles and make SDA functionality available to a broader audience. webSDA could be used as a tool for teaching Brownian dynamics or for experimentalists dealing with molecular interactions. webSDA was designed to run simulations with small molecules and/or short simulation runs. To run a simulation for a time period longer than 24 hours, the standalone SDA has to be used. In this case, the web server can be used to prepare the input for SDA, which is one of the major difficulties in using standalone SDA.
Workflow
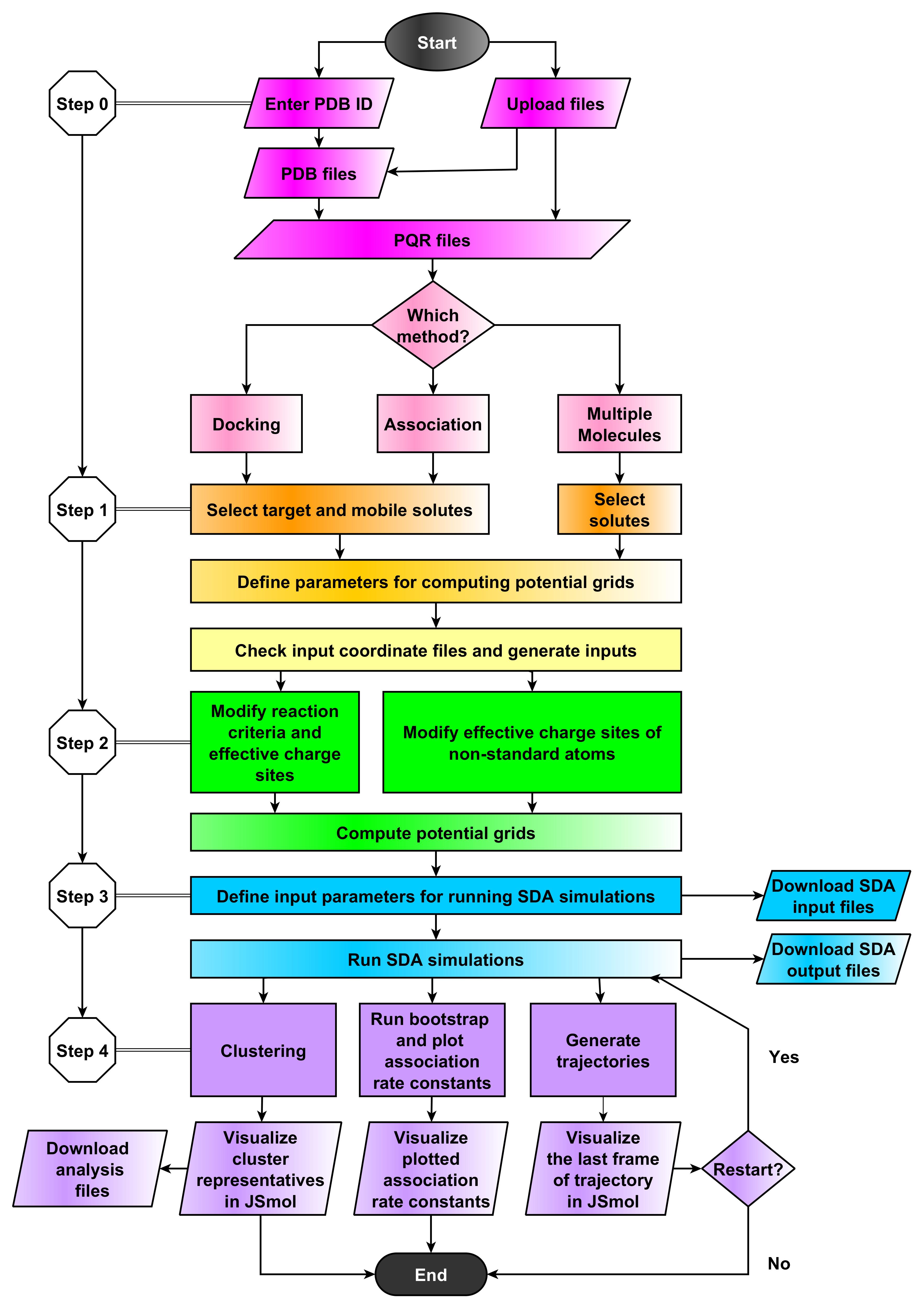
Frequently Asked Questions
For the computation of webSDA jobs, a cluster with 9 compute nodes with 48 cores on each node is used. The queuing system on the compute cluster uses the Terascale Open-source Resource and QUEue Manager (TORQUE) to manage submitted webSDA jobs. If the jobs exceed the current computational capacity, the jobs will be queued for the next available slot. For calculation of interaction grids, clustering, bootstrapping of association rate constants and generation of trajectories, 2 cores of one node are assigned to each job, whereas for SDA jobs, 8 cores of one node are assigned to each job. The maximum run-time of a job is 24 hours. For SDA jobs, output files are written and provided to users for the 24 hours that the job executed. For other types of job, no output files are generated if 24 hours are exceeded.
webSDA has been tested using Internet Explorer 11, Firefox 36.0.1 and Google Chrome 35. webSDA can be used with all browsers that support HTML5, Ajax, jQuery and JavaScript. Other browsers have not beed tested. The help links have been tested with Google Chrome and Internet Explorer, but for Firefox, an additional click of the URL in the newly opened tab is required to redirect to the correct section of the webpage.
The force field model used in webSDA is described in the appendix of Martinez et al. (2015)
and its cited references, and on the SDA 7 website.
The force field contains adjustable parameters in the electrostatic and hydrophobic desolvation terms,
which webSDA sets to reasonable default values.
Electrostatic desolvation grids are calculated in zero salt conditions, regardless of the
ionic strength used in solving the Poisson Boltzmann equation. This is done to allow the
use of an ionic strength independent value of parameter α in the potential term (see
Eqn. A4 in Martinez et al. (2015)),
as described in
Gabdoulline and Wade (2009).
Prior to December 2015, webSDA used the ionic strength dependent approach described in
Gabdoulline and Wade (2001).
As electrostatic desolvation generally makes a small contribution to the overall interaction
free energy between solutes, any differences due to this choice are assumed to be small.
The hydrophobic desolvation potential contains a parameter β that defines the
proportionality between the hydrophobic desolvation energy difference that occurs when two
solutes form a binding interface, and the total loss of solute surface area on binding. In
webSDA a value of -0.013 kcal/mol·Å2 is used. This value has been chosen as it
has been shown (Gabdoulline and Wade (2009)) to
give a good balance between accuracy and computational efficiency in docking and association
calculations. In multiple molecule simulations, a larger magnitude of up to
-0.019 kcal/mol·Å2, may give a better agreement to experimental results.
Benchmarks for SDA have been computed (Martinez et al, submitted). webSDA supports
“SDA docking” and “SDA association” jobs with up to 20000 SDA runs and “SDA multiple molecules”
simulations of up to 120000 ps. If users want to run longer SDA jobs longer, they have
to download and use the standalone SDA 7.
For the examples given in webSDA, for “SDA docking” and “SDA association”, 200 SDA
runs take less than one minute, 2000 SDA runs take ca. 2 minutes and 20000 SDA runs take ca. 20 minutes.
For the “SDA multiple molecules” example, a 2000 ps simulation takes ca. 1 minute and 20000 ps simulation
takes ca. 3 minutes.
For interaction grid calculations, webSDA is able to process files
that are up to the memory limit on the compute cluster (64 GB). As an example, the nucleosome (ca.
24000 atoms), it takes about 10 minutes to generate the grids.
webSDA uses the PDB2PQR software (version 2.0.0, http://www.poissonboltzmann.org/)
to convert PDB files to PQR files. In this step, all water
molecules in PDB files are deleted.
One limitation of
this step is that, currently webSDA DOES NOT
support the PDB2PQR conversion of ligands and cofactors. So if
there are ligands or cofactors in PDB files and they are
important for interactions of solutes, we strongly recommend
users to generate their own PQR files with ligands or cofactors
and upload these PQR files to webSDA.
First, check if you have the "NTR" or "CTR" problem below.
Check if all the records of the non-protein residues (ligands
and waters (HOH or WAT)) have been changed "ATOM" to "HETATM",
please delete all the waters that are not needed.
If
there are other problems, please do not hesitate to contact us.
First, make sure that the chain ids are present and correct.
Delete the "CTR" and "NTR" specification from the pqr file (should not be needed anymore).
If you test with a pqr file generated with WHATIF, please let us know the results.
The python script will try to add effective charge sites for
this residue/ligand if its net charge is different from 0.
Some empirical rules are applied:
- If it is a residue (ATOM record), the effective charge sites are assigned to the oxygen atoms
- If it is a ligand (HETATM record), one unique effective
charge site with the net charge of the ligand is assigned to
the atom closest to the center of geometry of the ligand.
The add_atoms file will contain all the atoms of the ligand, so that you can modify the effective charge sites in "Step 2".
The diffusion coefficients are computed with the dcc
(beta version...link not updated) tool provided in SDA 7. It is
a fast and simple algorithm (which still need to evaluated). In
order to use reliable input, we advise the use of the HYDROPRO
software.
The correct information is needed in the case
of:
- association rates, these parameters enter in the final evaluation of the association rates (will influence the absolute values of the rates)
- sdamm, it modifies the dynamics of the solutes and (not implemented yet in webSDA:) influences the (post-)calculation of effective ( compared to self ) translational and rotational diffusion coefficients
Distance (Å) = (MaxDim1 + MaxDim2)/2 + 12. MaxDim1 is the maximal value of the x, y and z dimensions of the static solute and MaxDim2 is that of the mobile solute.
Make sure that the provided pdb files represents a correct bound structure: no overlap between the solutes and a minimum of 3-4 donor-acceptor pairs (hydrogen bonds).
First, two distances for the two solutes are computed:
- Dist1 (Å) = ( √ 3 * MaxGrid1 + MaxDim2 ) / 2. MaxGrid1 is the maximal grid dimension of the static solute and MaxDim2 is the maximal value of the x, y and z dimensions of the mobile solute.
- Dist2 (Å) = ( √ 3 * MaxGrid2 + MaxDim1 ) / 2. MaxGrid2 is the maximal grid dimension of the mobile solute and MaxDim2 is the maximal value of the x, y and z dimensions of the static solute.
- start_pos (Å) = Max(dist1, dist2) + 10
- c_value (Å) = start_pos * 2
- swd1 (Å) = MaxDim1 + MaxDim2 + 15, MaxDim1 and MaxDim2 are the same as mentioned above.
- swd2 (Å) = swd1 + dt2 - dt1, dt1 is the minimum timestep (default 1 ps) and dt2 is the maximum timestep (default 20 ps). Both dt1 and dt2 can be modified by the users in the third step of webSDA.
The script expects a PQR file (See Limitations) and proceeds as follow:
- Check the residues have a correct total charge (integer value)
- Check if the provided structure contains protein and/or DNA sequences
- Try to set N and C terminal accordingly to the PQR file (NTR/CTR/NTN/CTN)
- Add missing ions, ligands or unknown charged residues to the add_atoms file
- Generate a PDB file in the format expected by SDA (pqrnoh_X.pdb)
- Compute geometric properties (dimension, atom at the center of geometry of the solute,...)
- A Debye-Huckel approximation is used to determine the APBS grid size.
- Generate a shell script to compute: APBS electrostatic grid, effective charges and other grids
Output files:
- A general output containing warnings (program still continues) and errors (program stops with an exit code of 1)
- A specific log file (PrepSDA.log) which reports all info, warning, error (used by webSDA)
Main limitations:
- The format of the PQR file is relatively strict:
- All ligands and ions must be specified with the HETATM keyword
- In case of a bound ligand (e.g.: HEME in CYP), HETATM must be used for the full residue to get a correct selection of add_atoms
- Chain ids must be present (it is an option in the python script, not set by default in webSDA)
Links
webSDA uses information from a variety of third party tools.
Scientific Software
Software development frameworks and tools
![]() Imprint
Privacy
Project
Terms and Conditions
Citation
Copyright © 2014-2015 by HITS gGmbH. All rights reserved.
Imprint
Privacy
Project
Terms and Conditions
Citation
Copyright © 2014-2015 by HITS gGmbH. All rights reserved.