webSDA example: docking of barnase and barstar
The "SDA docking" module is used to generate structures of the diffusional encounter complex of two input solutes. Multiple conformations of one solute can be treated by using multiple PQR files. Docked complexes can be required to satisfy biochemical constraints from experimental data or sequence analysis. This is done by defining reaction criteria that must be satisfied for an encounter complex to be recorded. In this example, barnase and barstar are docked, webSDA reproduces the binding mode of the crystal structure.
Step 0: upload the PQR files of barnase and barstar
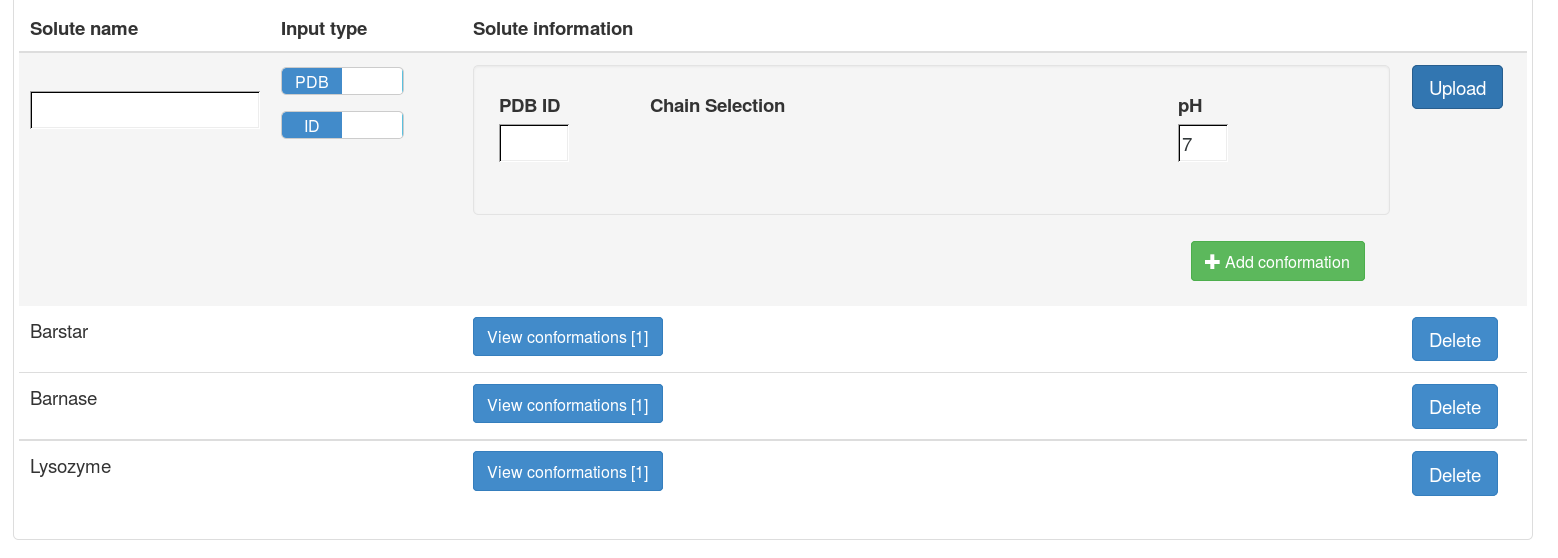
After uploading each file, the information on the uploaded solute will be shown on the right side of the file upload panel, including the name of the solute and the number of PQR files for this solute (number of conformations). A solute can only be deleted when it is not associated with any projects.
Now, choose the "SDA docking" method:
This goes to the page to set up a new docking project.
Step 1: choose solutes for the docking project
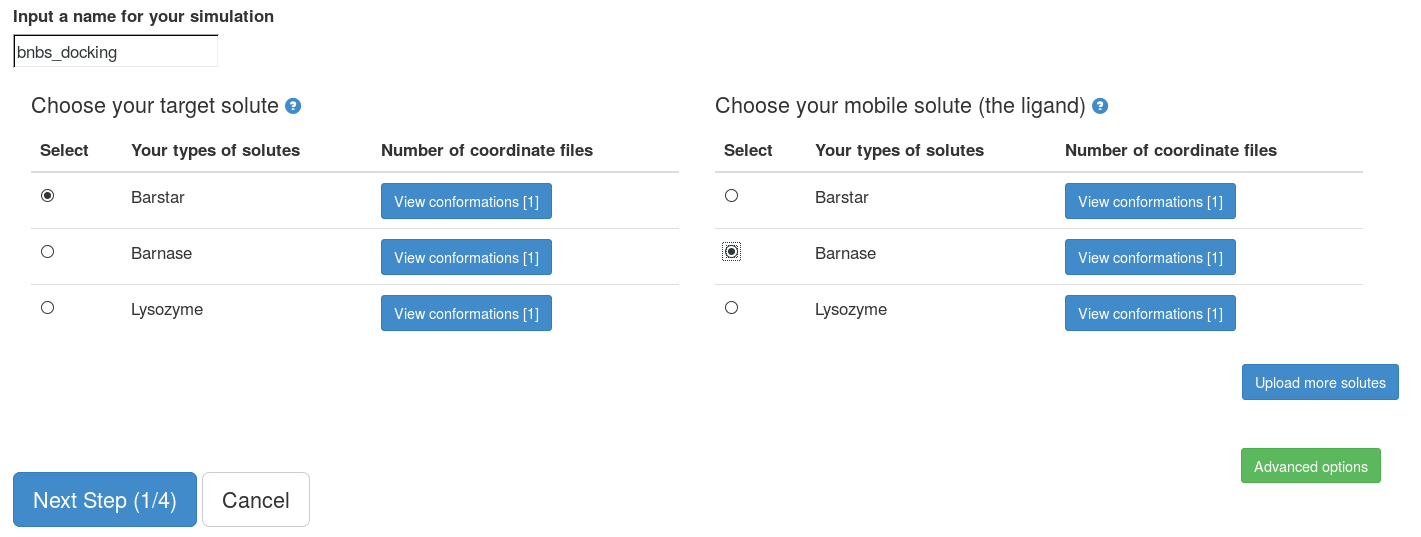
Here, we choose "Barnase" as our target solute and "Barstar" as our mobile solute (the ligand). NOTE: It is recommended to choose the larger solute as the target solute, this makes SDA simulations much faster. We use all default parameters in the "Advanced options".
A python script in the background checks the PQR files, generates necessary information (net charge, center of geometry, molecular weight etc.) and input files (to calculate the interaction terms).
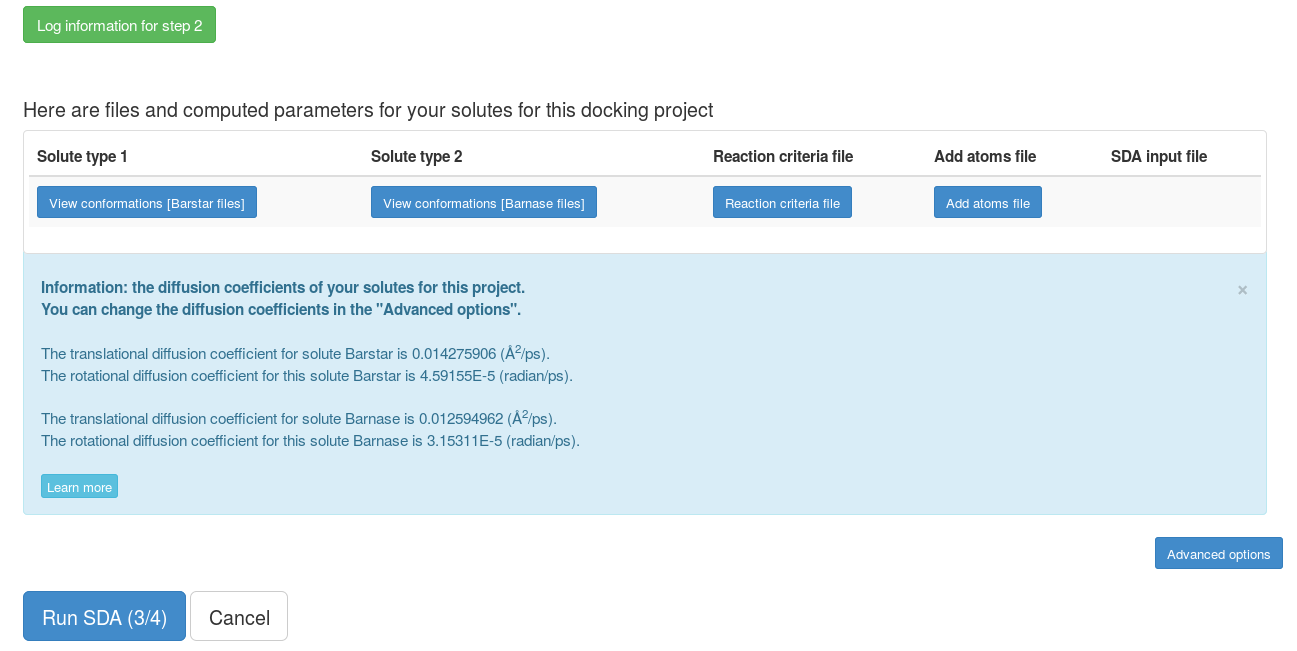
Step 2
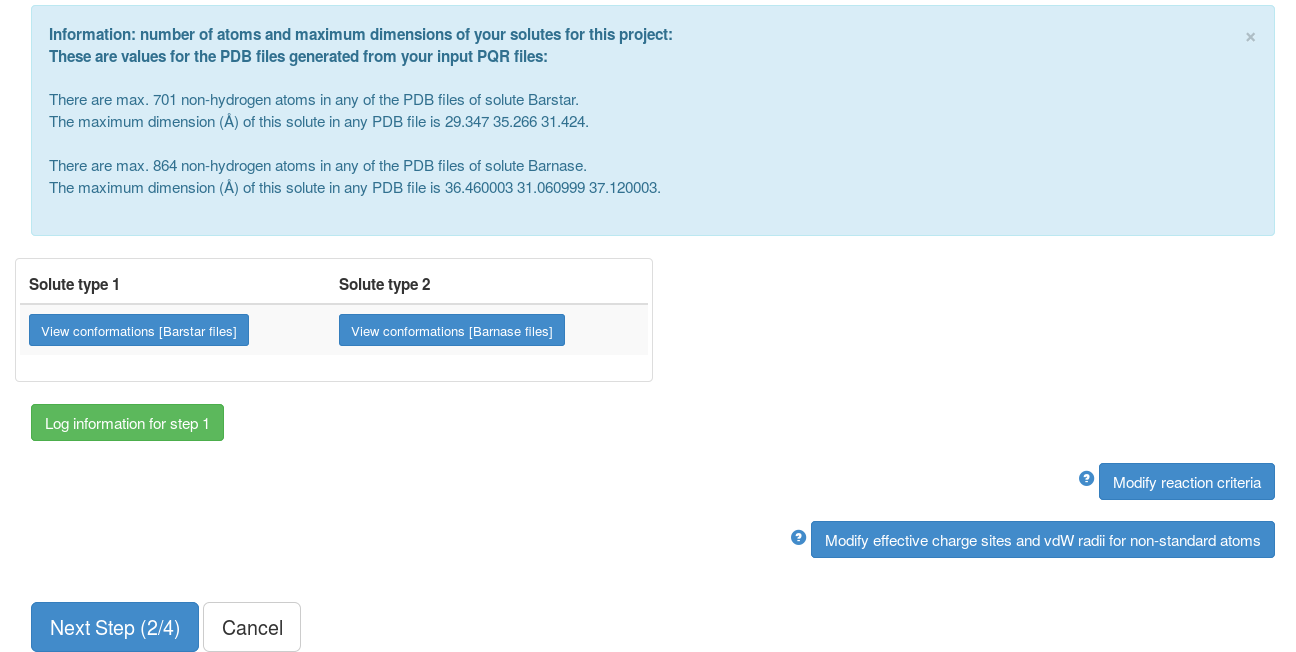
This page shows some of the information from the previous step, the reaction criteria defined (when to record complexes) and the non-standard atoms whose effective charges sites (normally on ligands) are not known. The reaction criteria, effective charge sites and VdW radii can be modified but here we use default values. After submission, the grid files for the interaction terms (here only electrostatic interaction forces) will be computed for each solute.
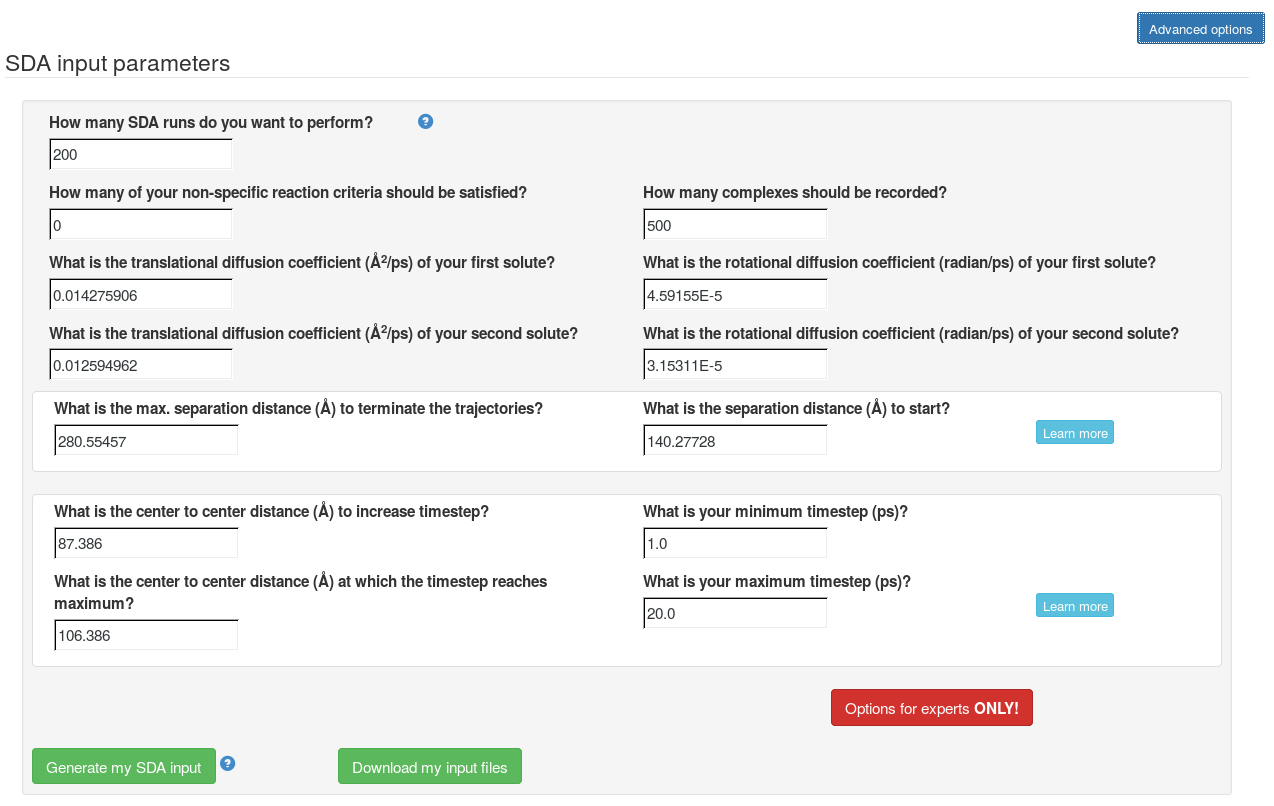
Step 3: Define input parameters
Now, the grid files for computing the interaction forces have been generated. In the "Advanced options", the SDA input file can be generated and all the files can be downloaded to run SDA7 standalone. The web server can only be used to run short SDA jobs. To get more accurate results, these input files should be used to with standalone SDA7 for a greater number of SDA runs (e.g. 5000).
We submit the SDA job with default parameters. For this barnase and barstar docking run, it takes about 2 minutes.
Step 4: Cluster the recorded complexes

The SDA log file gives detailed information about the SDA run. The "complexes" file contains all the complexes recorded. This file gives the position (rotation and translation) of the two solutes. The next step is to cluster the complexes and to write the coordinates the representative of each cluster to a PDB file so that you can analyze your results.
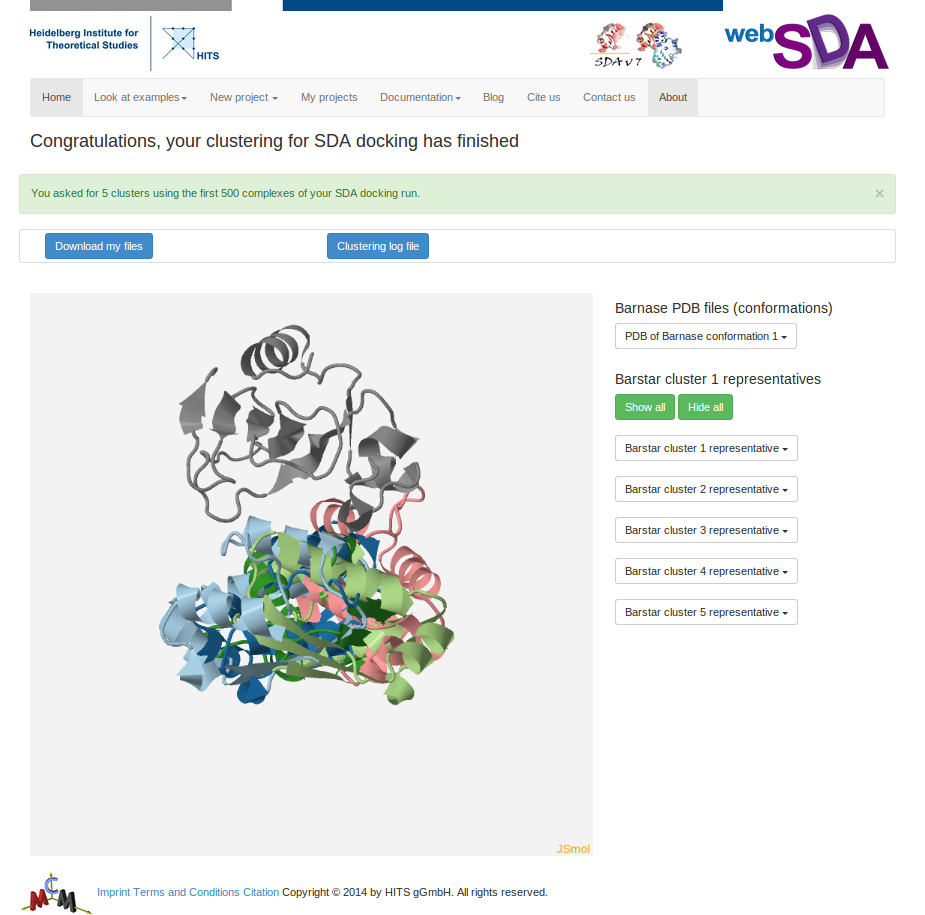
This page shows the target solute and the cluster representatives of the mobile solute in JSmol.
![]() Imprint
Privacy
Project
Terms and Conditions
Citation
Copyright © 2014-2015 by HITS gGmbH. All rights reserved.
Imprint
Privacy
Project
Terms and Conditions
Citation
Copyright © 2014-2015 by HITS gGmbH. All rights reserved.